
Induced Pluripotent Stem Cells for Drug Screening in Sickle Cell Anemia
Approximately 100,000 Americans have sickle cell disease; worldwide, ~300,000 affected children are born each year. Sickle hemoglobin polymerization drives the pathophysiology of this disease. HbF, the predominant in utero hemoglobin that is replaced early in life by adult
hemoglobin, inhibits sickle hemoglobin polymerization. One therapeutic goal is to develop drugs that can induce a high concentration of HbF in sickle erythrocytes, therein protecting the cell from polymer-induced injury. Increasing HbF levels to ~10 pg in 70% to 80% of sickle erythrocytes should have substantial clinical benefit. This is rarely accomplished with hydroxyurea, the only approved agent that attacks the
basic pathophysiology of disease. The development of novel, small molecule therapeutics for the disorder has been hampered, in part, by
the use of animal models and transformed human cell lines for drug discovery and validation that do not faithfully recapitulate the complexity of the disease and result in a high failure rate in human trials.
These observations form the basis for our central hypothesis: Patient-specific iPSCs will revolutionize pre-clinical drug screening for sickle cell anemia by allowing for the testing of novel inducers of HbF expression against the genetic variations found in a diverse patient population. Using a flexible, iPSC- based system capable of recapitulating ‘adult-type’ blood cell production, we will perform studies to validate this hypothesis as summarized in Fig. 1 and below:
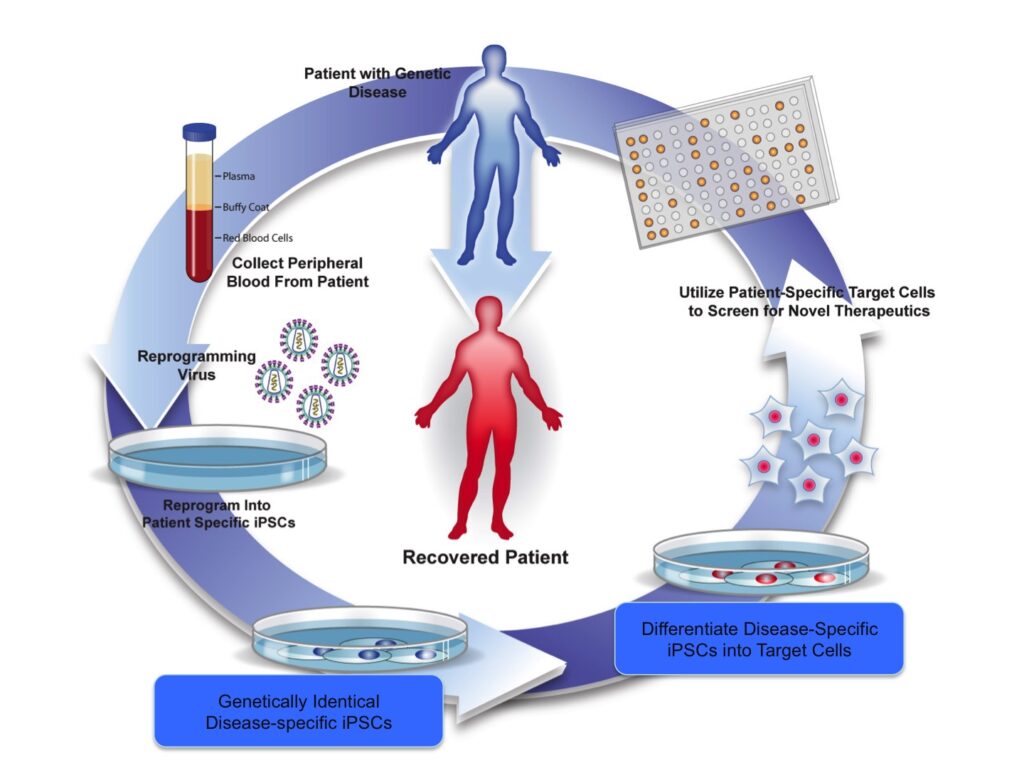
Objective: Harness our iPSC-based platform for the development and validation of novel therapeutics for sickle cell disease. A specialized directed differentiation strategy coupled with our library of ethnically diverse, sickle cell disease-specific iPSCs will be used to create a flexible platform for the development and testing of novel therapeutics. Sickle iPSC-derived erythroblasts will be used to screen the Boston University Center for Molecular Discovery’s (BU-CMD) library of structurally complex, bioactive small molecules for efficacy in increasing HbF in an ethnically diverse patient population.
The proposed studies use novel systems and strategies to: 1) identify new, small molecule HbF inducers, 2) give insight into the basic biology behind globin switching during blood cell development, and 3) lay the groundwork for the clinical development and testing of novel agents or pathways involved in the treatment of sickle cell disease.